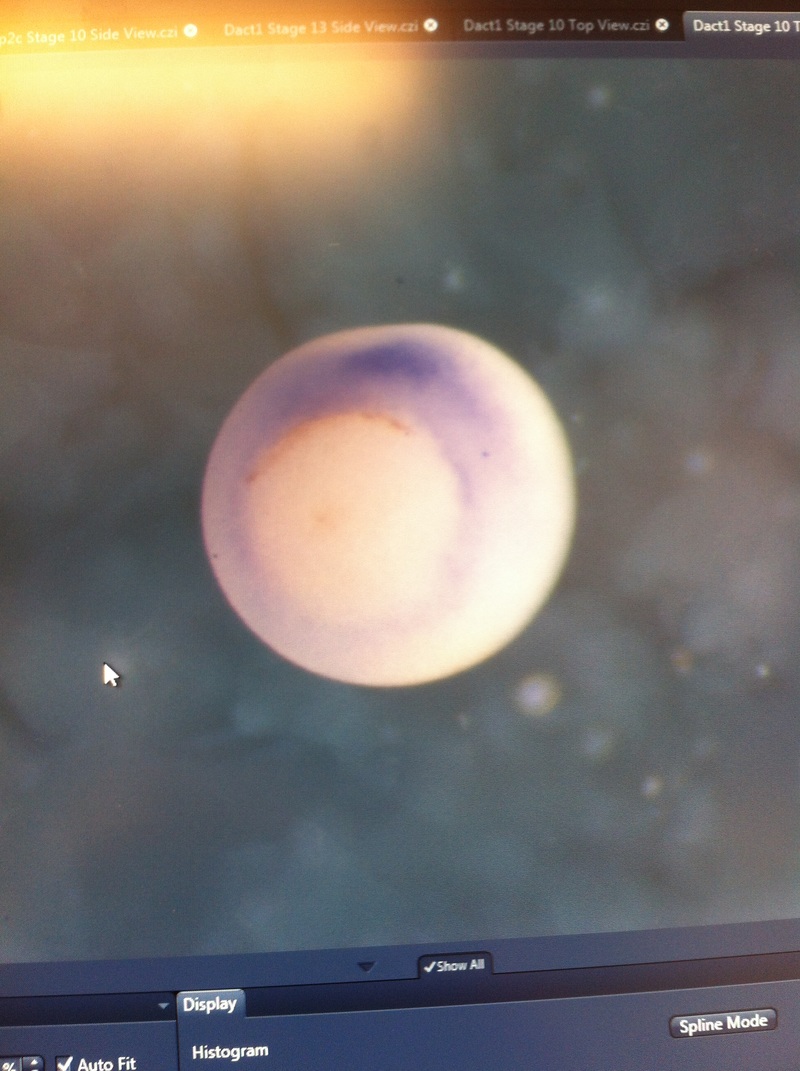
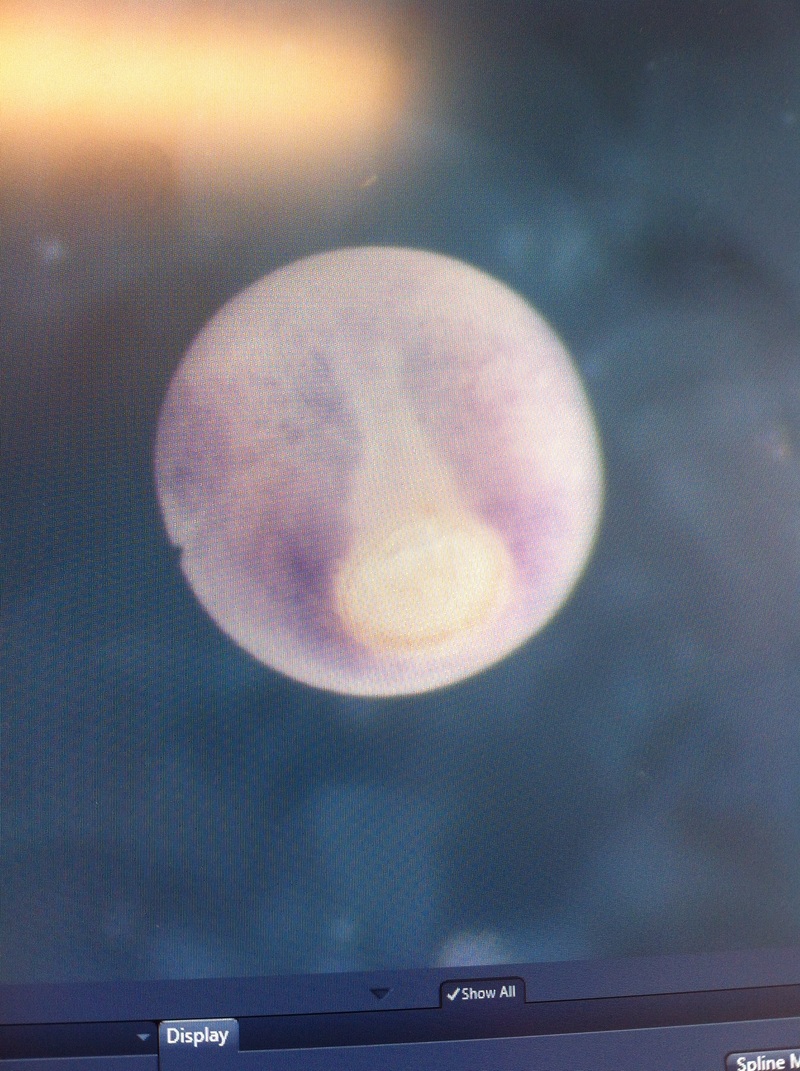
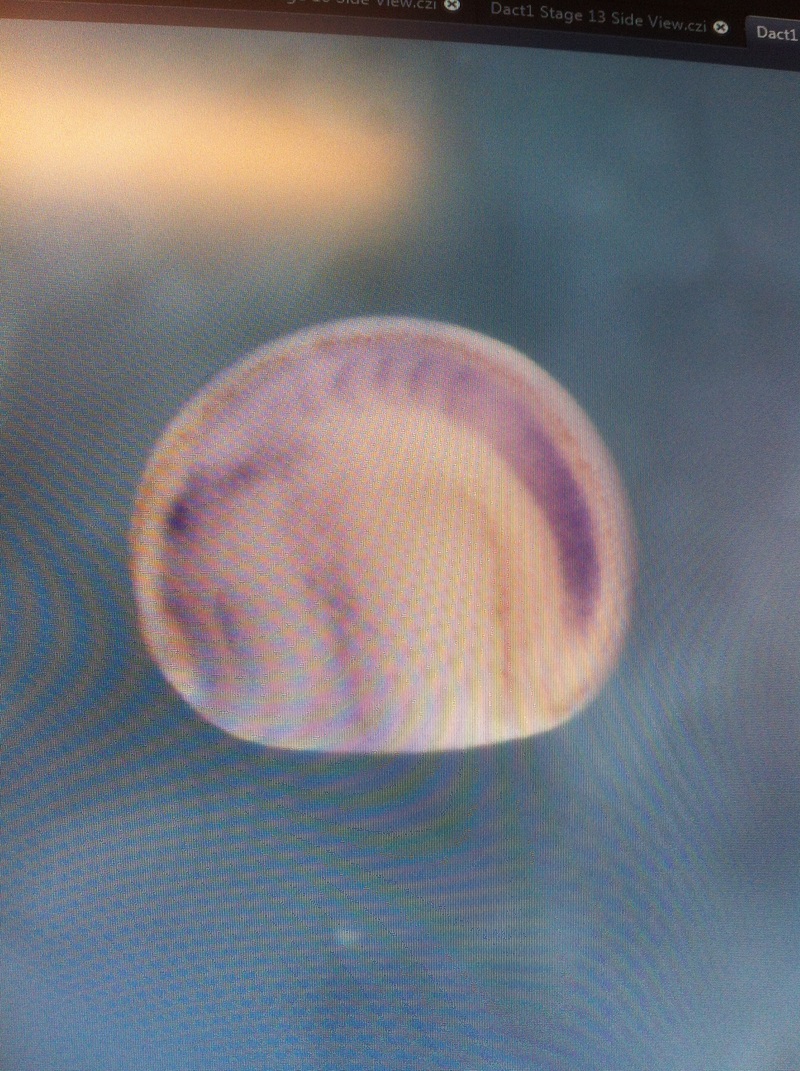
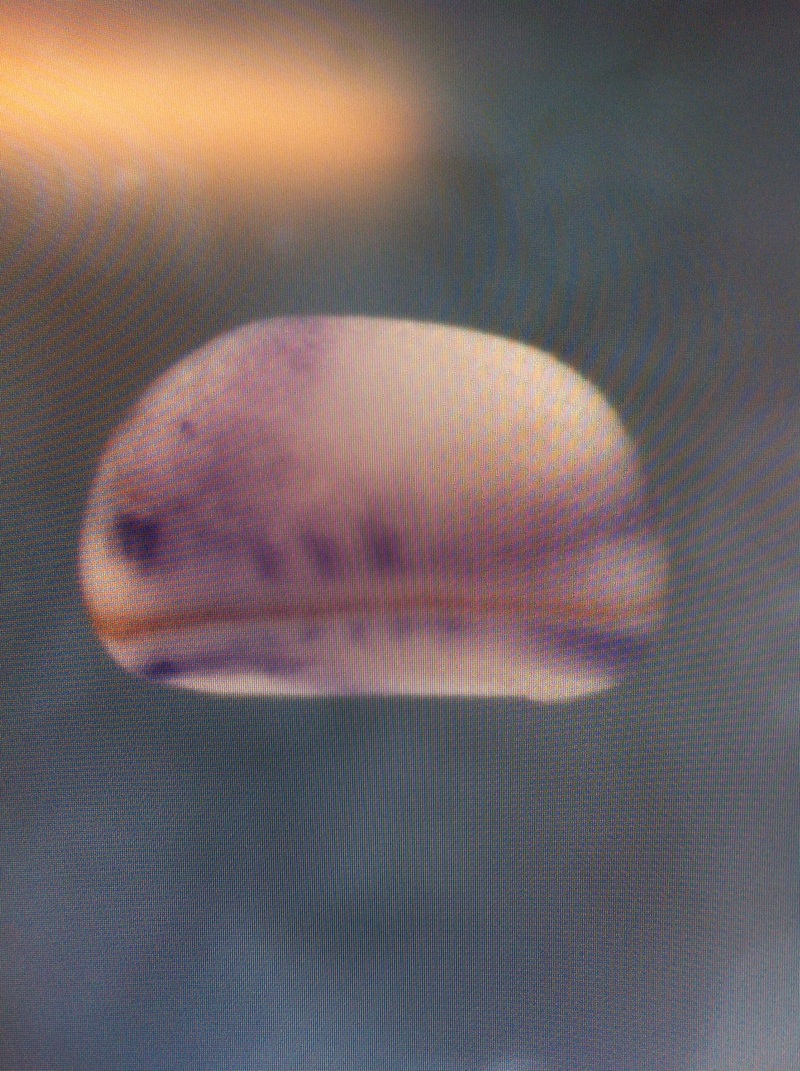
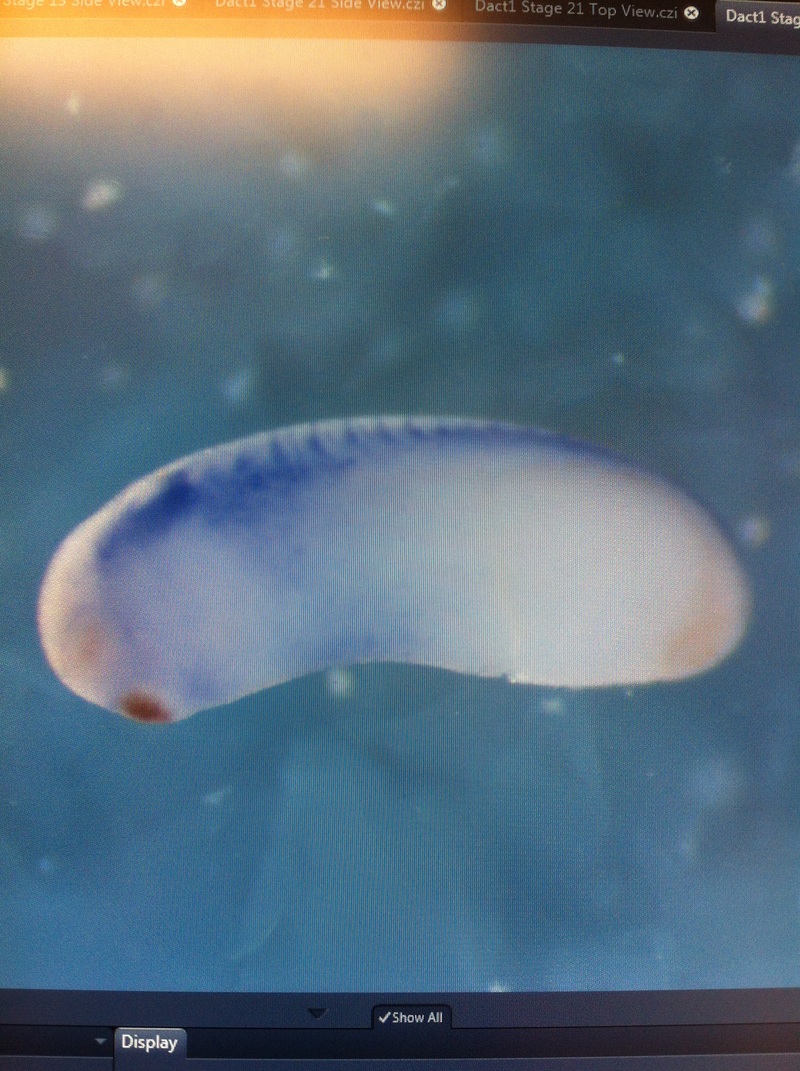
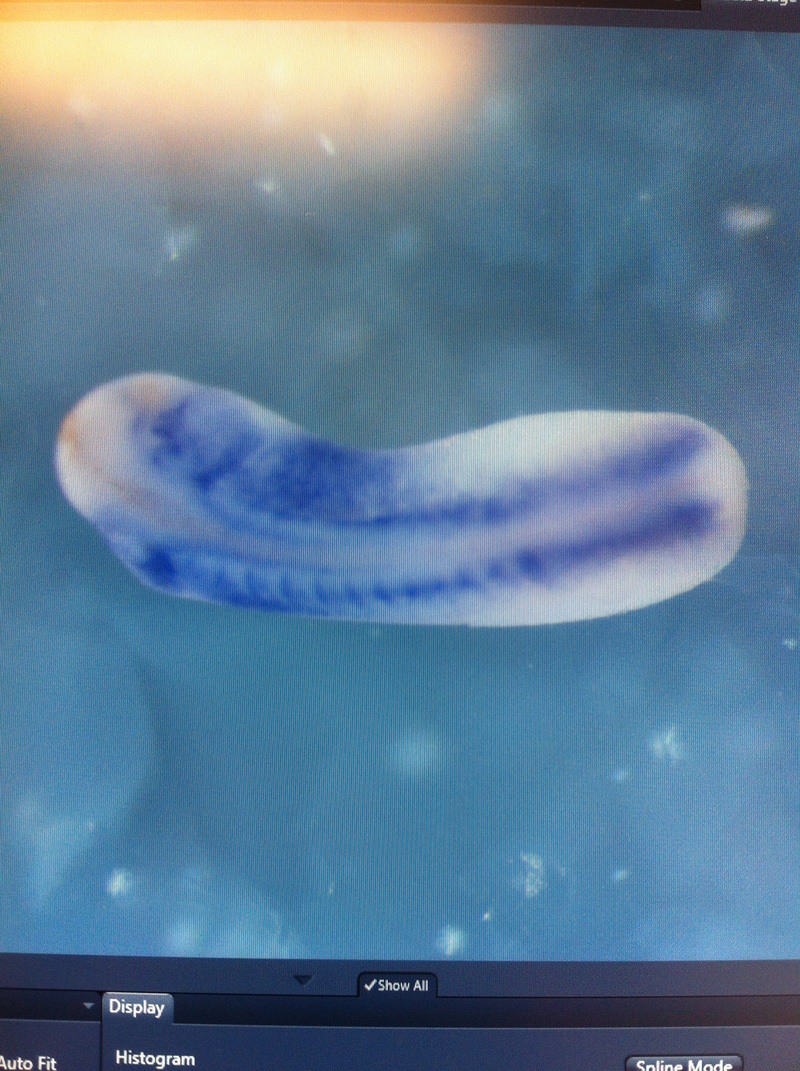
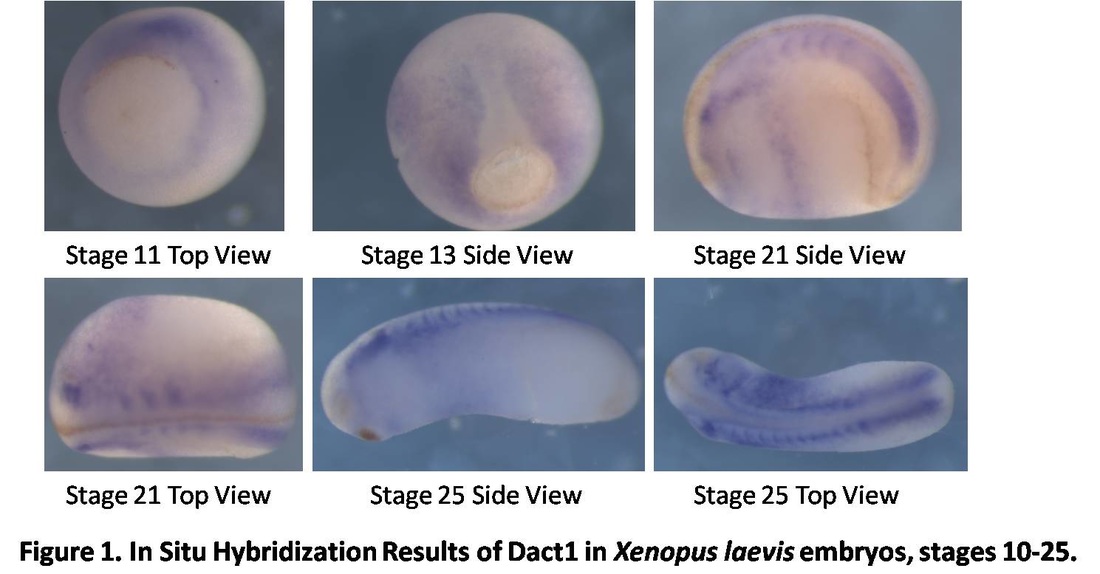
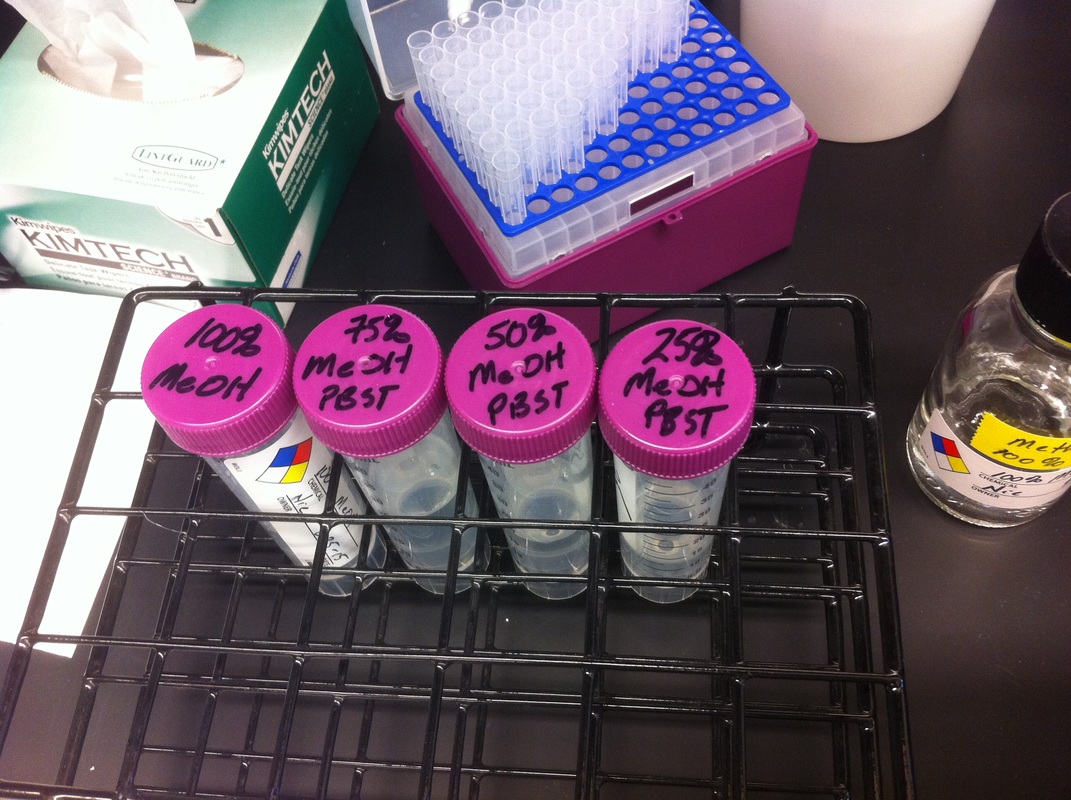
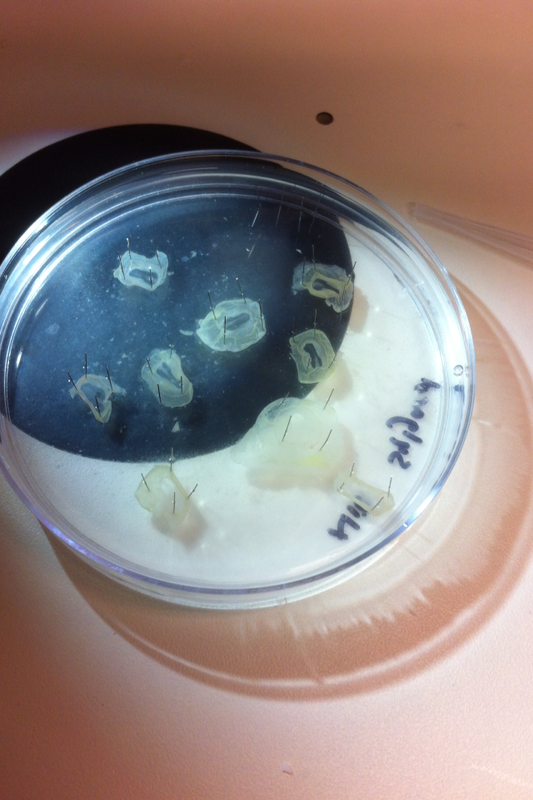
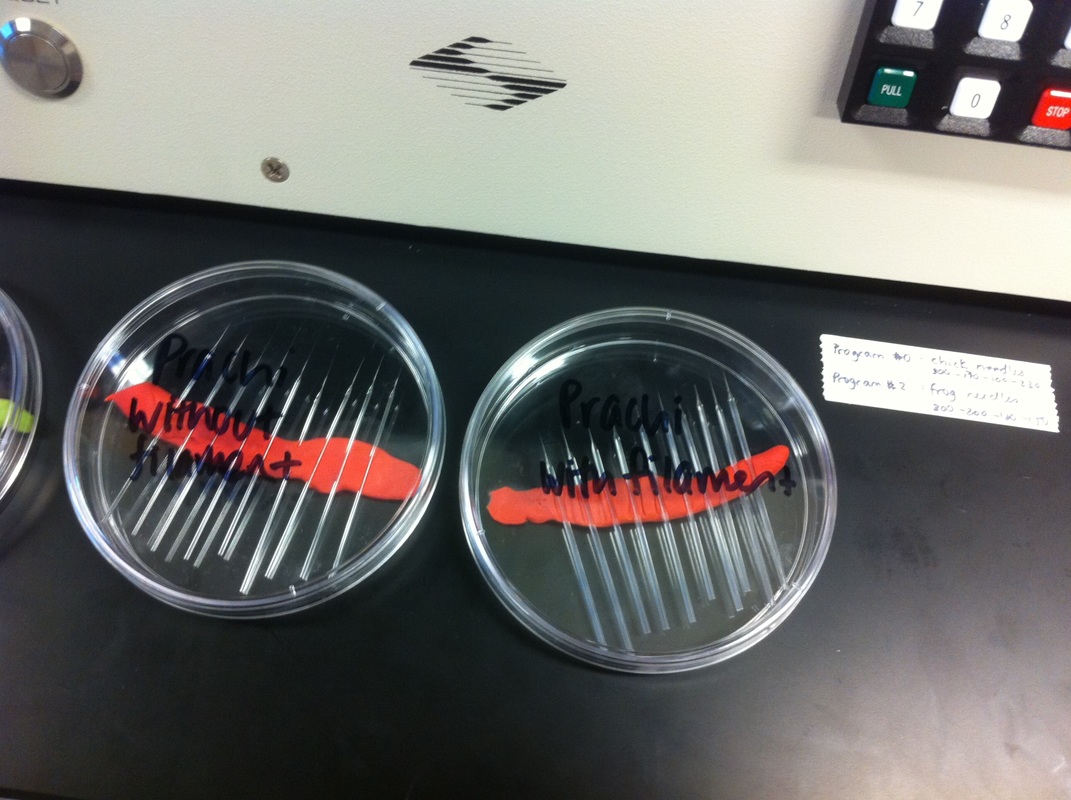
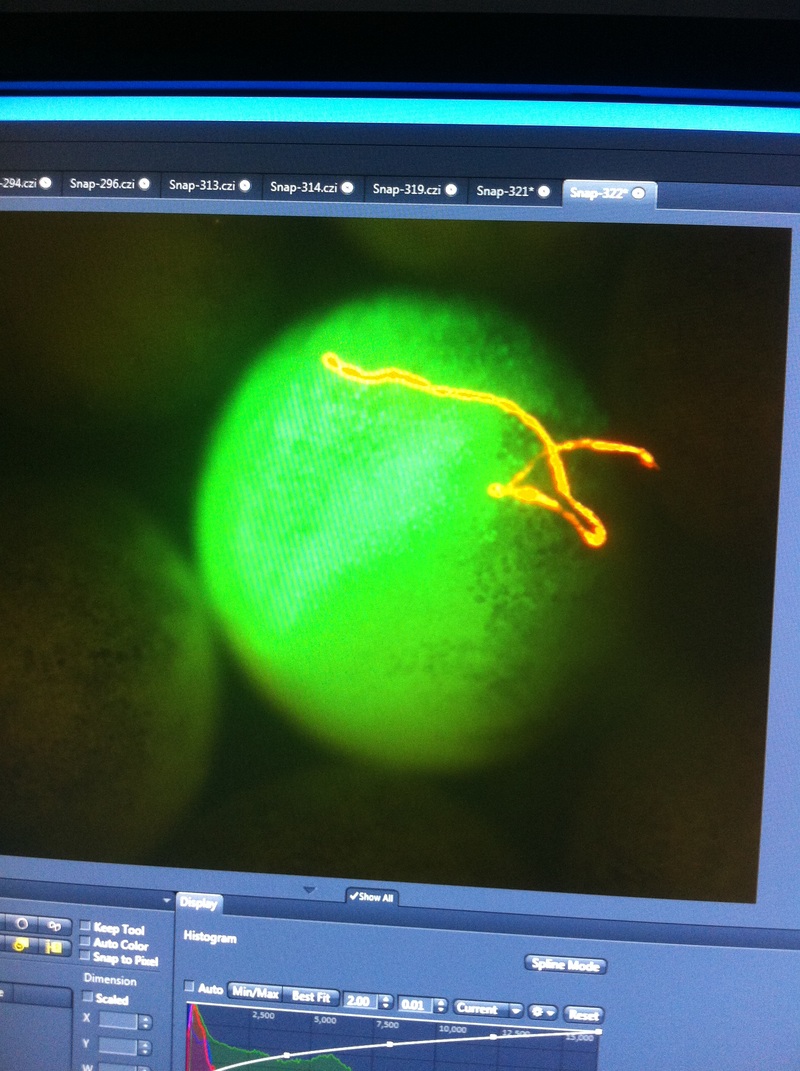
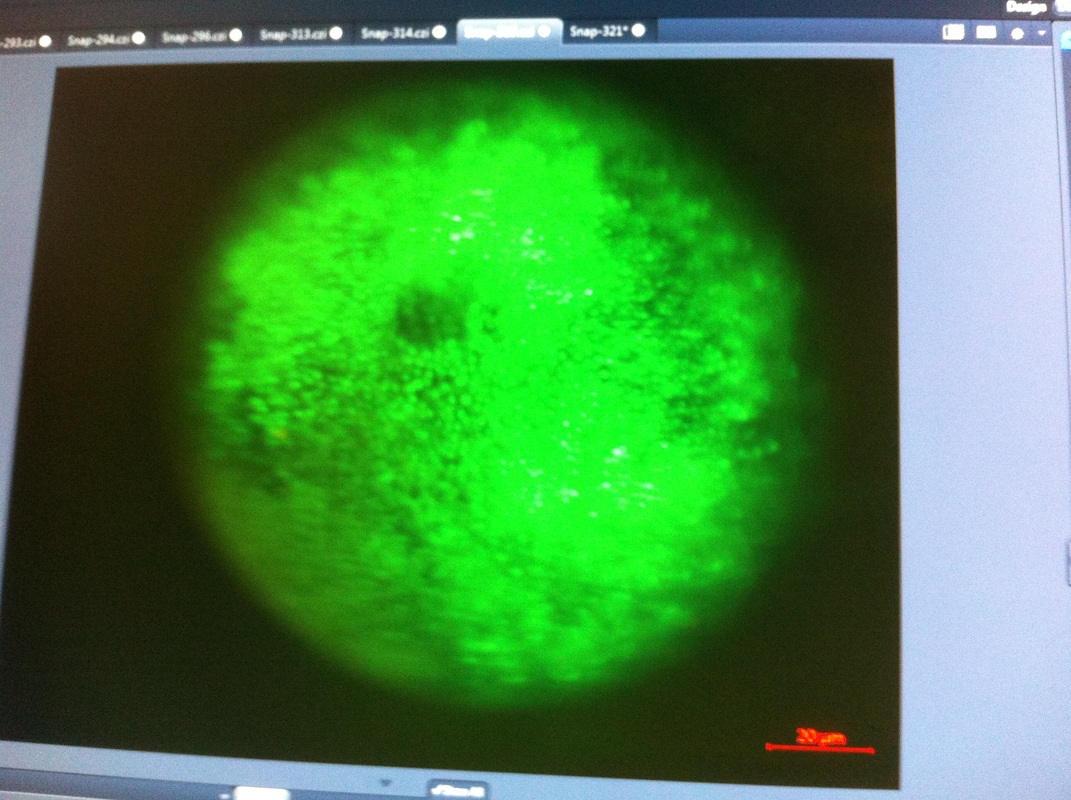
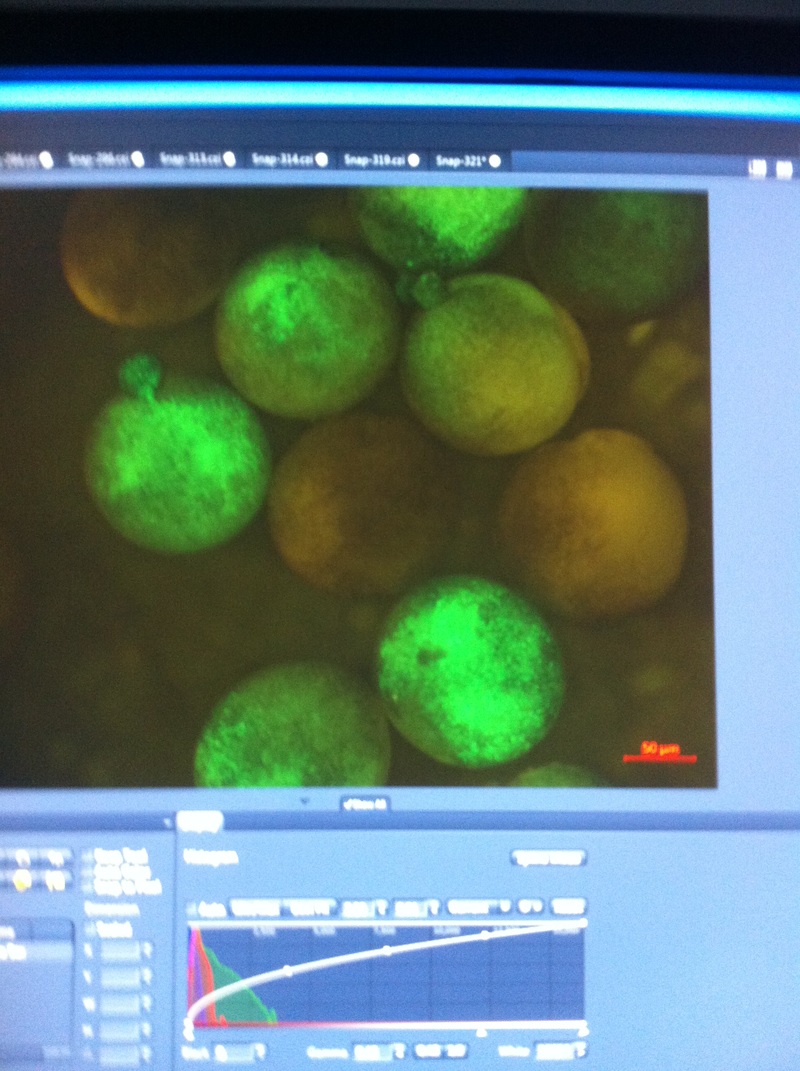
Over the course of the semester, I have been fixing frog embryos and learning the skills of injecting them, but for the very first time, Toby, Dahee, and I performed our very first whole mount in situ hybridization protocol! First we separated four different stages into 12 vials, with 4 vials each for Toby, Dahee, and I. We placed approximately 30 embryos in each vial, through stages 11, 13, 21, and 25. Then, we begin Day 1 of the In Situ. We begin with the rehydration steps: First with a 100% Methanol wash, 75% Methanol & 25% Water wash, 50% Methanol & 50% Water wash, 25% Methanol & 75% PTw, and finally, a 100% PTw wash, each for 5 minutes. Then we wash all the vials again with three PTw washes for 5 minutes each. Afterwards, we carry out the Proteinase K treatment for approximately 5 minutes. Then, we rinse the vials with two 0.1M triethanolamine washes for 5 minutes each. We conduct three acetic anhydride additions, each for 5 minutes. Then we wash twice with PTw for 5 minutes each. The vials are then refixed for 20 minutes in 4% paraformaldehyde in PTw. Afterwards, we wash off the excess paraformaldehyde with five PTw washes for 5 minutes each. The vials are then prehybdrizied overnight at 60 degrees Celsius. Then, we replace with 0.5 mL RNA probe solutions with our respective vials, approximately 1 microgram/milliliter per probe, which is then hybridized overnight at 60 degrees Celsius. The next day, the probe is removed and is replaced with hybridization buffer, the vials are then incubated at 60 degrees Celsius for 10 minutes. Three SSC washes are conducted at 60 degrees Celsius for five minutes each. Another 30 minute SSC wash occurs at 37 degrees Celsius, followed by two more SSC washes at 10 minutes and 30 minutes respectively, at 60 degrees Celsius. The vials are then washed twice in MAB for 10 minutes each. Another wash with MAB in 2% BMB Blocking solution occurs for one hours, followed by an MAB in 2% BMB Blocking Solution Reagent and Ab (anti-dioxygenin AP antibody) overnight at 4 degrees Celsius. The last day of the in situ started with 5 washes in MAB for 1 hour each. The embryos are then washed twice with Alkaline phosphatase buffer for 5 minutes each. The last wash is replaced with BM Purple, and the in situ has come to a conclusion! Attached below are pictures of the in situ hybridization with my Dact1 RNA Probe. The captions on the pictures indicate the type of picture and the stage of the embryos.


































 RSS Feed
RSS Feed
